



In the highly regulated world of medical device manufacturing, the relentless requirement to minimize and mitigate risk places a huge onus on design, development, and manufacturing teams.
That’s why companies, large and small, are looking for software solutions to help ensure consistency and objectivity in the creation, auditing, and monitoring of critical risk management processes.
There is a lot of complexity—that’s a given. So how can medical device manufacturers be sure the tools they’re using are fit for the purpose? Here are three key questions they should be asking.
No. 1: What's Wrong with Tried and Tested Tools Such as Excel?
There's nothing intrinsically wrong with basic tools for projects that are relatively simple. For many processes, such as basic engineering processes, these are fine.
But medical devices are radically more complex. As a result, it’s trickier to use tools like Excel spreadsheets to manage all of this. In fact, using an unstructured tool like Excel—designed for organizing and analyzing numbers—causes errors, an overwhelming verification effort, and potential challenges from regulatory authorities. This can have serious effects such as delays to market, rejections, post-market problems and complaints, even potential lawsuits.
Once you introduce multiple projects and multiple variants, including risk scoring and a range of other parameters that sometimes change depending on modifications to regulations or new guidance, the potential for errors becomes very real. At best this can delay commercialization. At worst, this could create a flawed process or product with dangerous outcomes.
With complex medical devices, compliance and risk management become proportionally complex. Manufacturers can’t rely on multiple spreadsheets. Copy and paste doesn’t cut it. That's why companies are turning to structured software solutions that can substantially reduce the administrative burden and eliminate the potential of errors such as mistyping or subjective scoring of risk data.
No. 2: What Do I Need to Consider When Developing a Reliable Risk Management Process?
Companies use a comprehensive list of questions to improve their risk analyses and management in order to meet demanding regulatory compliance requirements.
As we know, each relevant regulatory authority will focus on product safety. This involves identifying risk from many different perspectives, including all of the potential ways someone could be harmed. That could be a surgeon, nurse, patient—in fact, anyone involved in the normal use or potential anticipated misuse. The device manufacturer must analyze hazardous situations, hazards, and harms including mitigation activities to create a device with high efficacy and low acceptable risks.
We can visualize this through a typical number of products, such as a pacemaker, or a surgical robot for kidney surgery, or a device that works with knee joints, or fusion pumps for diabetes treatment. In other words, medical devices that may be iterative, and almost certainly are based on innovation in technology or process—or both.
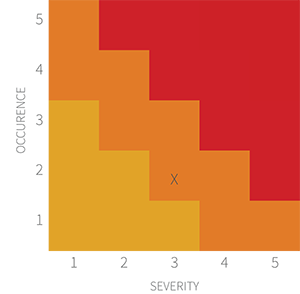 Potential questions include:
Potential questions include:
- What could go wrong with the device?
- What's the probability of occurrence?
- How severe is the harm?
- Is it a nuisance or a danger?
- Is this potentially fatal?
- What can we do to reduce the likelihood of harm occurring?
In each country or region, regulations can differ. The US is different from the EU, the UK, and Japan. Basically, there are different ways to submit information. The EU Regulations for medical devices (MDR) changed the rules on how to submit information. FDA rule changes have also required an overhaul of processes and submissions, something we refer to as regulatory churn.
Additionally, there are a number of industry standards to factor in, such as ISO:14971, which covers risk management for medical devices, as well as IEC 62366 (usability in human factors risk); IEC 60812 (which deals with Failure Modes Effect Analysis (FMEA); and ISO: 24971 (guidance on the application of ISO 14971), adding further complexity to the risk management.
That’s not the end of the story. Many larger organizations set their own processes and procedures and those also need to be factored into compliance and risk procedures and processes.
All of that means a powerful risk tool is needed to provide these core elements. But they need to be flexible enough to allow customers to fine-tune—hence the need to form reusable templates that companies can customize.
But it’s not just large organizations that must play by the rules. Smaller companies also need to navigate the complexities of risk management. They too need a scalable solution that’s fit for purpose, complete with risk tools that need to provide a comprehensive set of templates.
No. 3: Why Should I Trust You and Your Tool?
It almost goes without saying that all medical device companies should gauge their risk management tool provider’s level of industry knowledge and competency. When it comes to understanding this complex industry, with all of its changing compliance implications, you need a dependable partner.
But risk management is not exclusive to the medical device industry. Knowing and understanding the context and what terms like “risk,” “harm,” “hazard,” and “mitigation” mean in this industry is critical.
This itself demands a process that is not relying on subjectivity, but is peer-reviewed, date-stamped, and provides an audit trail. The parameters can be verified by accessing an online library of all types of hazards, depending on the device application.
People and spreadsheet data cannot provide the assurance of consistency, and that’s a key reason that basic data systems aren't fit to deliver the compliance data demanded by regulatory authorities. In order to grant approvals, regulators need to review the evidence that everything is covered, that the structured data makes sense, and corroborates the potential risk in the right way.
Doing all of this increases credibility. This is not just about getting approval, but as with any regulatory system, human subjectivity matters. By showing the relationships between data, the manufacturer is able to demonstrate that a robust and thorough process is in place. It is evident that you are serious about doing this right.
Risk cannot be assessed or managed in a vacuum. It needs to be considered in the context of requirements of the system and testing to harness consistent and accurate information.
Spreadsheets have their place, but for risk management and compliance of complex medical devices, they should not be the first choice for companies that need to get to market quickly and safely.
See how a comprehensive risk management tool can help you get to market faster with less risk!

