



Imagine developing a medical device or drug that could be claimed to work without any evidence….
And where profits could occur with unknown risk to users.
This was the reality before the FDA was established.
Through successive decades of developing regulation, the current standards for quality systems and design controls were realized. Proof of safe and effective use became the foundation of product design. However, today’s regulations still only provide minimum guidelines for quality-based design and best practices for manufacturing. Medical device manufacturers use the processes and procedures outlined by the quality system regulations to ensure that their medical devices are manufactured to consistently meet the design and intended use. One of the crucial elements of the FDA quality system regulation is the subsection on design controls, which guide manufacturers on planning, design definition, review, verification, validation, transfer to manufacturing, control of changes, and maintenance of the design history. Design controls are the keystone to the FDA’s enforcement of medical device regulations. In fact, almost 60% of the 4,000 citations issued in the past decade are related to design control.
Medical Device Design Control Regulations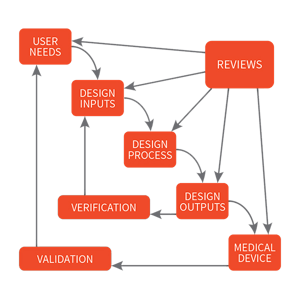
The term design controls originates from the FDA standard 21 CFR 820.30, which is part of the overall Quality System Regulation defined by the FDA. As stated in 21 CFR 820.30, “Each manufacturer of any class III or class II [and certain class I devices]... shall establish and maintain procedures to control the design of the device in order to ensure that specified design requirements are met.”
On a global level, the concept of design controls, as outlined by FDA 21 CFR 820.30, is related to the product realization as defined within ISO 13485:2016 standard, in particular, part section 7.3, “Design and Development”. Though these are closely related one significant difference between the two is that ISO suggests a more risk-based approach to a company’s quality management system (more on this later).
Compliance to ISO 13485 is used to support the general safety and performance requirements of the forthcoming EU MDR. The FDA is in the process of aligning 21 CFR 820.30 with the ISO 13485 standard. Therefore, a global medical device manufacturer that is planning to sell in the EU, US, and other countries is wise to use ISO 13485 as a basis for developing their processes and procedures.
Best Practices for Design
Clearly Define User Needs and Design Requirements
Design inputs, which include user needs and technical design requirements, are the foundation for design. By clearly defining these from the beginning in a way that is complete, clear, concise, and measurable you set up your design process for success. If a need or requirement is incomplete, ambiguous, or conflicting you risk the final product not satisfying the intended use. Another effect of inadequate requirements is unintended consequences when making changes to the product by not fully understanding the requirements. Measurable inputs are critical to determine whether you have satisfied the criteria. By taking the time upfront, to clearly and fully define your user needs and design requirements, you are setting your design process up for success.
For more information, read our blog post on The Importance of Linking User Needs to Design Requirements.
Use Evidenced-Based Rationale
The rationale behind a requirement should not only be clear but it should be evidenced-based (i.e., pullout-strength of an anchor based on biomechanical forces or its use related to bone density). Using solid logical rationale, helps ensure the intent of the design decision is clear for any proposed changes in the future as well as helps to justify the original design choice.
Say What You Do, And Do What You Say
At the end of the day, your design controls or design process are all about whether your end product meets your originally defined design and user needs. Therefore, you must follow your own written procedures, and ensure that your processes are defined such that any trained personnel can follow them and end up with the same result.
On a simplistic level, if your design calls for a two-inch round red sticker on your packaging, you must manufacture with a two-inch round red sticker on your packaging.
Take a Risk-Based Approach
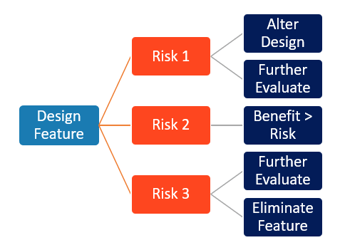
Risks are inherent throughout the product development process - risks exist and evolve as design features are changed or added to a product. Risk management must be a part of the overall design controls process so that potential risks or harms to people, property, or environment are considered and analyzed throughout the product’s development and lifecycle. Risk evolves with the design and manufacture; and should be frequently re-assessed and applied to all elements of design (definition through testing), manufacture, and post-market. Assessing risk in parallel with design, while concurrently creating trace matrices to show the interactions, enables the development team to see the full impact of choices and options.
Improve Your Design Process
FDA regulations and ISO standards can seem complicated, but your design process does not need to be. Compass, our SaaS solution, with our integrated approach to risk and design, can help you take control of your medical device development, and enable safer products to be brought to market faster, with less risk.
To learn more about our approach to Design, contact us or request a demo.
