



In February 2019, FDA published a final guidance document that expanded parts of its 510(k) premarket notification program. The guidance establishes a new pathway: The Safety and Performance Based 510(k). With this novel premarket pathway comes several new opportunities and benefits to device manufacturers, plus some challenges. Understanding these benefits and challenges together—as well as where the Safety and Performance 510(k) fits in the premarket environment—can help your organization better navigate your device’s path to regulatory submission.
Safety and Performance: An Abbreviated 510(k)
 FDA has three overarching categories of 510(k)s: Traditional, Special, and Abbreviated. The Special and Abbreviated methods are intended to further streamline review timelines and processes. The new Safety and Performance 510(k) falls under the Abbreviated pathway, meant for what FDA defines as “well understood” device types.
FDA has three overarching categories of 510(k)s: Traditional, Special, and Abbreviated. The Special and Abbreviated methods are intended to further streamline review timelines and processes. The new Safety and Performance 510(k) falls under the Abbreviated pathway, meant for what FDA defines as “well understood” device types.
The Abbreviated submission method differs from the Special and Traditional methods due to its reliance on established regulatory guidance, standards, and specific controls. Under the umbrella of the Abbreviated 510(k) program, the Safety and Performance pathway has a specific and limited scope. It additionally aligns with FDA’s substantial equivalence and least burdensome provisions.
Like other Abbreviated 510(k)s, the Safety and Performance pathway relies on guidance documents, special controls, and recognized standards for demonstrating conformance and substantial equivalence. It also leverages performance criteria as a basis for what must be submitted and reviewed by regulators.
The Importance of Performance Criteria
Performance criteria are established through FDA-recognized performance standards. These standards can cover everything from performance characteristics and quality processes to testing and labeling. According to 21 CFR 861, they can be established for:
- Class II devices
- Class III devices that have undergone reclassification to Class II
- Class III devices that, as a condition to premarket approval, must reduce or eliminate associated risks
Within this range are safety and performance criteria that apply to this new 510(k). According to the recent guidance, FDA expects submitters under the Safety and Performance method to:
“…use robust versions of those [Abbreviated 510(k)] mechanisms, which contain all the performance characteristics necessary to support a finding of substantial equivalence for a device type, rather than using direct predicate comparison testing to support a finding of substantial equivalence for some of the performance characteristics.”
Rather than requiring a direct comparison with a predicate device’s safety and effectiveness data, this pathway asks manufacturers to simply show their device performs at the same level of safety and effectiveness as those already cleared and legally marketed.
What Device Types are Eligible?
Currently, there is no published list of eligible device types for the Safety and Performance 510(k), but FDA anticipates its publication in the near future. However, the agency has established criteria for evaluating appropriate device submissions for the pathway, which include:
- An eligible predicate device cited in the submission
- Indication(s) for use/technological characteristics that do not raise different safety and effectiveness concerns
- Performance criteria aligned with one or more legally marketed devices (of the same type)
- Evidence the device meets all performance criteria identified by FDA
All of these criteria must be met; if not, submitters still have the option to submit under the Traditional, Special, or Abbreviated pathways.
Benefits of the Safety and Performance 510(k)
This novel method of premarket notification has the potential to be valuable for medical device manufacturers. It has three prospective benefits: the optimization of industry and regulatory tasks, streamlining of regulatory reviews, and improved response times/time to market.
Optimizing Effort
The Safety and Performance pathway may cut down on the burdens of preparing a traditional 510(k). Because FDA understands the device well—its intended use, indications for use, benefits to the patient population, etc.—additional work to establish that same data in a premarket submission is unnecessary. Through the Safety and Performance pathway, time and resources can be optimized for greater focus on critical compliance activities.
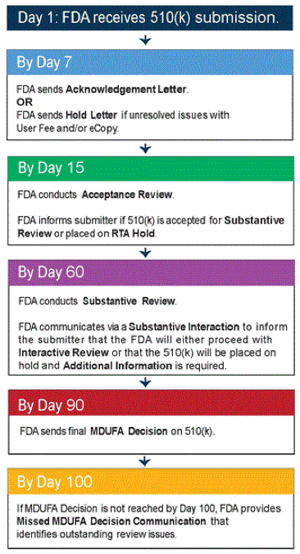 Streamlined Regulatory Reviews
Streamlined Regulatory Reviews
Because regulators only look at submission data related to conformity with performance criteria, less time needs to be spent in the review process. Reviewers only have to establish that the device submission is in compliance with established guidance documents, special controls, and recognized consensus standards. This allows FDA the ability to get more products to market that can positively impact patient outcomes faster—without compromising safety and efficacy.
Improved Response Time to Market/Stakeholders
Improved response time is a big possible benefit of the Safety and Performance 510(k)—especially for manufacturers releasing new and updated versions of existing products, and/or those looking to expand their product portfolios. Being able to address user needs quickly with a more streamlined pathway, or simply launching a low-cost, well-understood product into the market, can have long-term benefits for organizations of all sizes.
Challenges and FDA’s Future Direction
The biggest challenge facing the Safety and Performance 510(k) pathway is its scope. In the short term, manufacturers are limited to submitting a limited number of device types, still yet to be fully defined. However, it’s reasonable to anticipate this pathway will expand as it matures. Therefore, a wait and see approach may better serve medical device manufacturers in the present; those who are unsure about what the pathway can offer them could see greater opportunities open up to them in the near future.
