


Managing the risks of your life science product can be a complex task. Some risks must be assumed, while others need to be controlled, transferred, monitored, or designed out of the product entirely. Implementing risk controls to reduce and manage the impact of associated risks, as well as their probability of occurrence, is the most common approach manufacturers leverage in their design and development efforts.
In understanding how to identify, evaluate, and enact risk controls in your life science product development, there are a number of issues and approaches to take into account. From regulatory requirements and guidance to international standards and industry principles, the factors that influence risk controls are many.
FDA and Risk Control
FDA approaches thinking about risk controls for life science products through two avenues. The first is guidance documents recommending and outlining risk management principles for manufacturers to follow; the second is the product classification process itself.
FDA Guidance
 FDA’s current thinking on risk management is encapsulated in their guidance document Guidance for Industry: Q9 Quality Risk Management. They state, in no uncertain terms, that “The manufacturing and use of a […] product, including its components, necessarily entail some degree of risk.” FDA further goes on to explain their position on the purpose of the risk control approach: reducing a given risk to an “acceptable level.” However, they do not take a stance on what acceptable levels of risk look like, deferring instead to the life science manufacturer’s judgment.
FDA’s current thinking on risk management is encapsulated in their guidance document Guidance for Industry: Q9 Quality Risk Management. They state, in no uncertain terms, that “The manufacturing and use of a […] product, including its components, necessarily entail some degree of risk.” FDA further goes on to explain their position on the purpose of the risk control approach: reducing a given risk to an “acceptable level.” However, they do not take a stance on what acceptable levels of risk look like, deferring instead to the life science manufacturer’s judgment.
Product Classification
Especially for medical devices, the product classification process is risk-based. Whereas Class I devices, for example, present the least amount of risk to users and patients, any device categorized as Class III is thought of as high-risk. Safety and efficacy are the chief concerns in the classification process, and the resulting premarket submissions evidence required by FDA needs to demonstrate:
- Product benefits outweigh risks to target population (when adequate risk controls are applied)
- Results for target population are clinically significant
- Unreasonable risks have been designed out of the product (i.e. FDA’s inherent safety by design principle)
 There are many risk considerations that tie into this evidence, including the extent of probable harms and the probability and duration of harmful events.
There are many risk considerations that tie into this evidence, including the extent of probable harms and the probability and duration of harmful events.
With device classifications, FDA also ties in a number of required and/or recommended controls based on the product type and the level of risk it presents. There are general controls that apply to all devices (registration, labeling, quality system regulation/good manufacturing practices, etc.), and these act as the backbone of your premarket submissions. Beyond these, there are special controls, which include additional things like additional labeling requirements, performance standards, specialized testing, adherence to recognized consensus standards, and so on.
Product classification helps your organization identify areas where risk controls are necessary for regulatory compliance. Greater recognition and adherence to these controls can assure regulators that your product is safe and effective.
ISO 14971 and ALARP
In understanding what acceptable levels of impact, severity, and probability of occurrence for a given risk look like, one particular principle can be applied: the as low as reasonably practicable (ALARP) approach. This principle comes from ISO 14971:2007—the international standard for medical device risk management—and can be effective in identifying acceptability criteria. ALARP takes into account: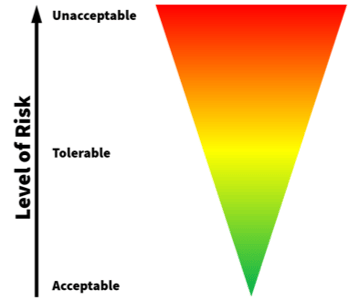
- Applicable national/regional regulations
- Relevant international standards
- The generally accepted “state of the art”
- Identified stakeholder concerns
ALARP refers to what level of control is most acceptable based on these and other factors. Again, the definition of what acceptable risk levels looks like are up to your organization to determine (14971 even explicitly states it does not specify acceptable risk levels), but these concerns are a good starting point.
Industry Tools for Risk Control Development
ALARP is a powerful approach in and of itself, but it can be applied in conjunction with other tools. Industry approaches such as the Triple Constraint (sometimes referred to as the Project Management Triangle) and the “innovation sweet spot”—also known as the Desirability-Feasibility-Viability Venn Diagram—evaluate proposed risk controls in the context of reasonable practicability.
The Triple Constraint
 Implementation of risk controls ultimately falls under the limitations of time, scope, and cost. These three factors of the Triple Constraint are important considerations for ALARP and risk control in general; evaluating the impact of these constraints on proposed risk controls provides practical insight into how feasible implementation is.
Implementation of risk controls ultimately falls under the limitations of time, scope, and cost. These three factors of the Triple Constraint are important considerations for ALARP and risk control in general; evaluating the impact of these constraints on proposed risk controls provides practical insight into how feasible implementation is.
For example, if a mitigation is broad in scope (the requirements and tasks necessary to realize that mitigation) and time-consuming to put into operation, but the resources necessary to support those two factors are not available, then the mitigation likely cannot be realized. Likewise, a project with proposed risk controls requiring significant resources and a broad scope but limited on time will struggle with incorporating controls.
Desirability-Feasibility-Viability Venn Diagram
The Triple Constraint is a potent tool not just because it supports ALARP, but also because it can roll into other tools. For example, it can be thought of as the “Feasibility” part of the Desirability-Feasibility-Viability Venn Diagram. This is a popular design thinking tool that acts as a framework for identifying optimal solutions for many different aspects of product development. Where these criteria overlap is what’s known as the “innovation sweet spot.”
This Venn diagram exercise is most effective after risk controls have been established, regardless of project stage. By framing proposed controls in context with these criteria, their value and necessity in the product design can be established and defined. Questions to ask regarding the criteria include: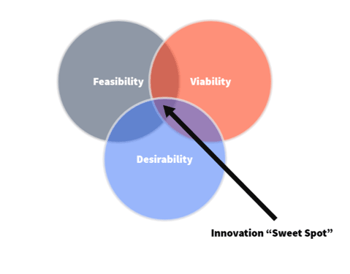
- Desirability: does the risk control fulfill user/patient needs? Could it impact user interactions negatively?
- Feasibility: is it possible to make use of the control without altering the product design? Does your organization have the time and technologies necessary for execution?
- Viability: can your organization afford to apply the risk control, and can payers afford your product with the risk control in place?
When asking these questions, keep in mind that some risk controls can’t be compromised to satisfy each criterion. However, this is actually a strength of using this framework. Understanding which controls are necessary helps your team find other areas to improve in your product design and in how the controls are engaged.
Why Risk Controls Matter
Risk controls are a fundamental part of your life science organization’s product development and compliance processes. Because life science products are regulated with a risk-based approach that prioritizes safety and effectiveness, controlling the risks your product presents to users and patients is necessary for approval and market access. How you identify, evaluate, refine, and enact risk controls into your product design has both regulatory and industry approaches to lean on. To best take advantage of these, understanding them early on can have positive impacts in getting your product from premarket to postmarket environments.
