



In March 2019, FDA’s Center for Devices and Radiological Health (CDRH) began their reorganization initiative first announced the year before. The Center has taken a major step forward in this process by launching the Office of Product Evaluation and Quality (OPEQ) in May 2019. With this new office, a number of important changes begin taking place at CDRH.
But what is OPEQ, how is it organized, and what effect will it have on your medical device organization? Here are eight important facts you need to know as OPEQ becomes fully operational.
1. OPEQ is a “super office”
Offices under CDRH’s purview are realigned as a result of the reorganization initiative. One of the consequences of these efforts is the establishment of OPEQ as a “super office”—an office that includes several functional units once separated in the previous CDRH structure. OPEQ combines the offices of:
- Compliance
- Device Evaluation
- Surveillance & Biometrics
- In Vitro Diagnostics & Radiological Health
2. It’s comprised of several health technology offices
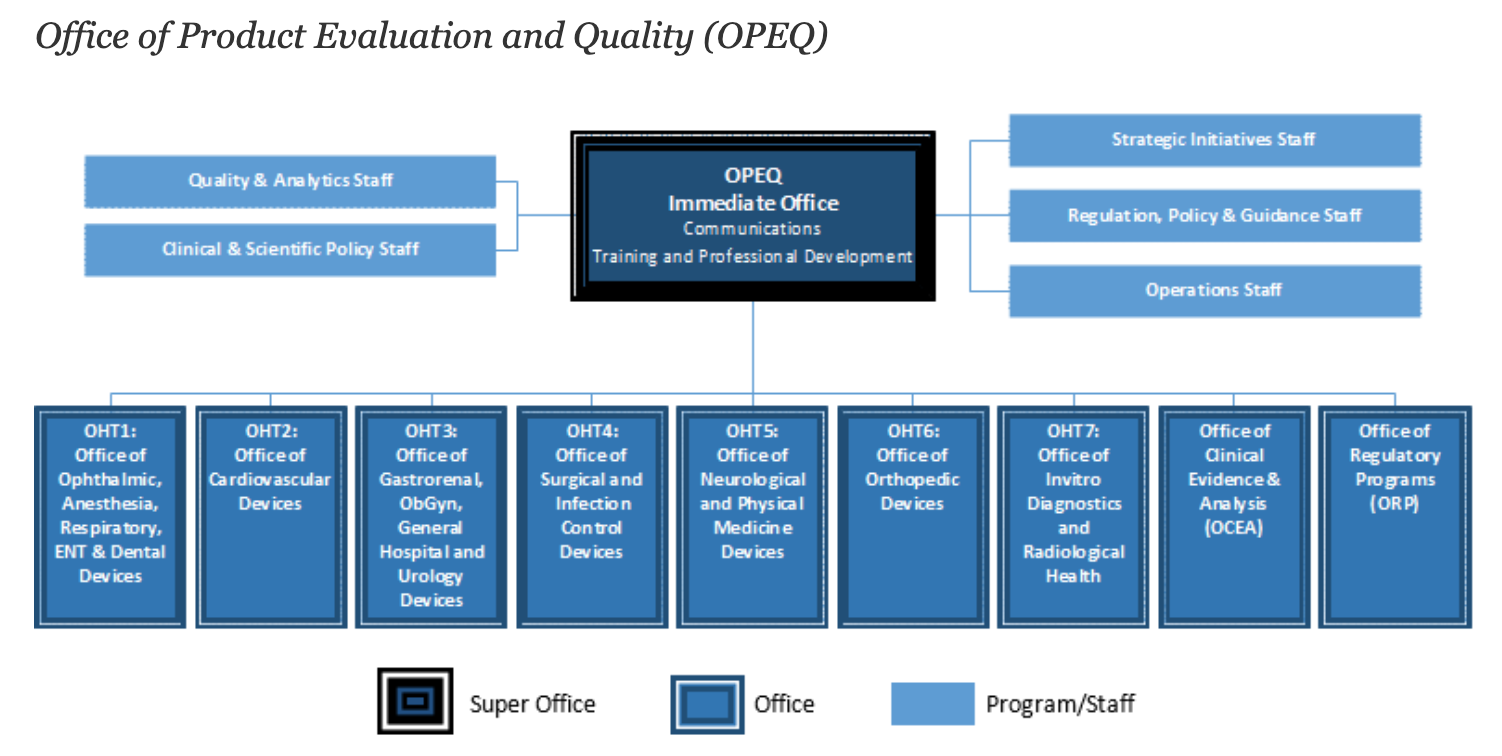 Overall, there are 11 separate offices that make up the overall OPEQ structure. Seven of these are Offices of Health Technology (OHTs) that focus on specific medical device types. These OHTs roughly line up with FDA’s device classification panels and encompass units for orthopedics, cardiovascular devices, in vitro and radiology, and others.
Overall, there are 11 separate offices that make up the overall OPEQ structure. Seven of these are Offices of Health Technology (OHTs) that focus on specific medical device types. These OHTs roughly line up with FDA’s device classification panels and encompass units for orthopedics, cardiovascular devices, in vitro and radiology, and others.
3. OHTs are broken into subdivisions
Each OHT is built of smaller working units. For example, OHT6: Orthopedic Devices includes divisions related to joint arthroplasty, spinal devices, and stereotaxic, trauma & restorative devices. This breakdown is likely reflective of FDA’s intent to make sure specialized medical devices can be adequately reviewed based on their respective panels.
4. OPEQ focuses on the total product life cycle
 The goal of OPEQ is to assure that consumers have access to safe and effective products throughout the total product life cycle (TPLC). This initiative covers both the premarket submission process for all device types and all categories of submission—510(k)s, PMAs, De Novo, Humanitarian Device Exemptions, and so on. OPEQ also oversees postmarket programs including recalls, audits, registrations, listings, and more.
The goal of OPEQ is to assure that consumers have access to safe and effective products throughout the total product life cycle (TPLC). This initiative covers both the premarket submission process for all device types and all categories of submission—510(k)s, PMAs, De Novo, Humanitarian Device Exemptions, and so on. OPEQ also oversees postmarket programs including recalls, audits, registrations, listings, and more.
5. The Office leverages a team-based approach
According to OPEQ Director William Maisel and CDRH Director Jeff Shuren, the new Office relies on a team-based approach. OPEQ’s aim is to encourage pre- and postmarket teams to work together when evaluating medical devices. With TPLC as the framework, this collaboration is intended to use as much data throughout a medical device’s life cycle to make regulatory decisions as informed as possible.
6. Your regulatory contacts may be impacted by OPEQ
Your medical device organization likely has contacts within CDRH fostered throughout the years. These relationships are important, both for you and for FDA. With the changes being made to the Center, however, some positions and authorities are likely to shift. To keep up with these potential changes, CDRH offers a management directory for manufacturers to use as necessary.
7. The Office oversees clinical laboratory regulations
 OPEQ has been designated to administer the Clinical Laboratory Improvement Amendments (CLIA). These regulations govern how laboratory testing is conducted and the certifications necessary for lab operation. While the amendments are jointly addressed by multiple agencies—FDA, the Center for Medicaid Services, and the Center for Disease Control—OPEQ takes on FDA’s roles in evaluating and reviewing laboratory testing.
OPEQ has been designated to administer the Clinical Laboratory Improvement Amendments (CLIA). These regulations govern how laboratory testing is conducted and the certifications necessary for lab operation. While the amendments are jointly addressed by multiple agencies—FDA, the Center for Medicaid Services, and the Center for Disease Control—OPEQ takes on FDA’s roles in evaluating and reviewing laboratory testing.
8. Clinical evidence is also a big focus
The new CDRH office has its own division devoted to clinical evidence. OPEQ’s Office of Clinical Evidence and Analysis (OCEA) supports policies and programs regarding clinical considerations such as:
- Clinical trials
- Biostatistics
- Epidemiological analyses
OCEA provides support for the other OHTs in reviewing devices that require expert clinical investigation and analysis of real-world data. In addition, this unit works with external stakeholders such as hospitals, healthcare professionals, and others to continue development of clinical evidence guidance and support.
